
Research projects
My research activity is focused on the engineering and the photophysical studies of fluorescent proteins. In particular, I'm interested by phototransformable fluorescent proteins that can either be activated from a non-fluorescent to a fluorescent state (photoactivatable FPs) or photoconverted from a fluorescent color to another (photoconvertible FPs) or reversible photoswitched between fluorescent and non fluorescent states (reversibly switchable FPs).
These proteins are very valuable for single molecule localisation microscopy techniques but their photophysics and structure details must be deeply understood to allow the creation of future better performing markers for super-resolution imaging.
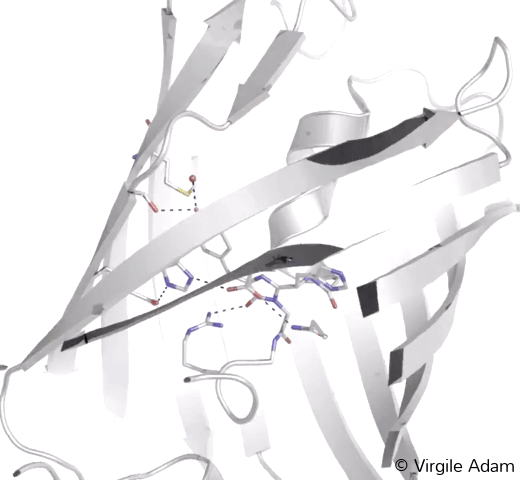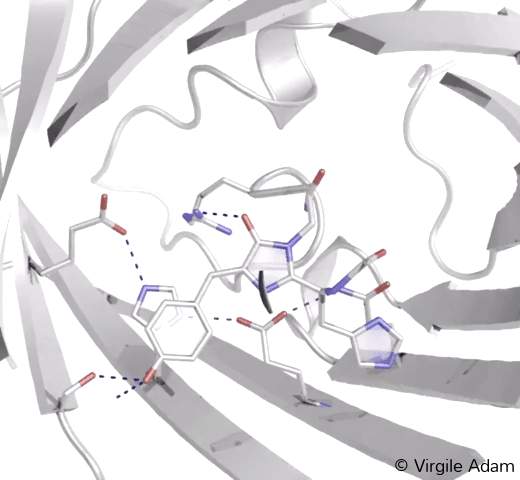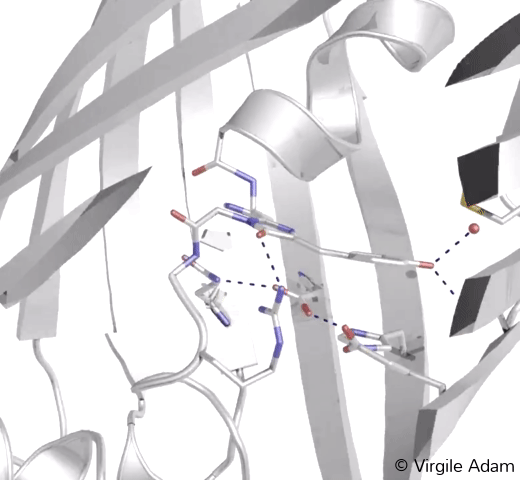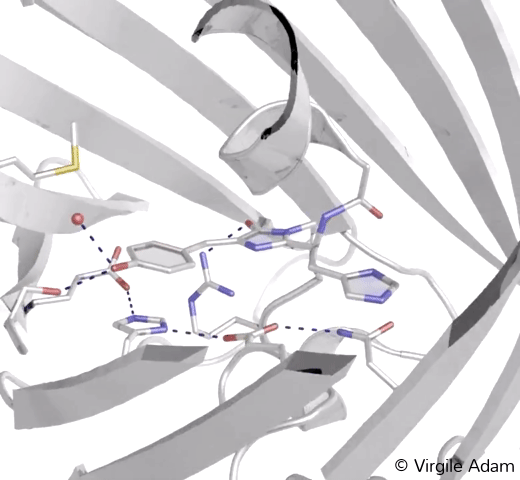

Different types of phototransformable fluorescent proteins (PTFPs) are shown on this image along with representative examples.
Among them: photoactivatable fluorescent FP (PAFPs) are capable of irreversible activation by light (generally near-UV) from a non-emisssive state to a fluorescent state.
Photoconvertible fluorescent proteins (PCFPs) can be irreversibly activate by light (generally near-UV) from a fluorescent form to another fluorescent form emitting with a weaker energy (red-shifted fluorescence)
Reversibly switachable fluorescent proteins (RSFPs) can be reversibly commutated between non-fluorescent an fluorescent states using two different excitation wavelengths, either starting from a stable off-state (negative RSFPs) or from a stable on-state (positive RSFPs).
The combination of both photoconversion and reversibly switching leads to the creation of biphotochromic fluorescent proteins.

How do fluorescent proteins change their colour?
Fluorescent proteins of the GFP family are the object of intense study due to their inherent bioluminescence, and have proved to be excellent markers for cellular imaging. In the last few years, "photoactivatable" fluorescent proteins have been developed whose fluorescence properties change as a function of their illumination conditions.
These proteins are crucial to the new "super-resolution" fluorescence imaging methods that permit images of living cells to be obtained at nanometre resolution. One of the most popular photoactivatable proteins in nanoscopy is EosFP. This protein normally fluoresces in green, but when illuminated with violet light its fluorescence changes to red. This "photoconversion" process involves the rupture of the peptide chain next to the chromophore and formation of an enlarged conjugated system, but its mechanism has proved elusive.
3D structure of the chromophoric environment of an FP in its green state. 2Fo-Fc electron density map is shown, contoured at 1σ
3D structure of the chromophoric environment of an FP in its photoconverted red state. 2Fo-Fc electron density map is shown, contoured at 1σ
Starting from X-ray crystallographic structures of the green and red forms of EosFP, we have employed QC/MM simulation methods to investigate possible reaction pathways and have been able to propose a mechanism for the photoconversion. Absorption of a violet photon by the protein promotes it to its first singlet excited state.
Approximately once in a thousand times, the singlet undergoes a forbidden transition to a triplet state. Once there, a proton transfer occurs that leads to a cascade of events that results in rupture of the peptide backbone, elongation of the conjugated system and red fluorescence.
This work is an advance in our understanding of the function of photoactivatable fluorescent proteins, and could allow the development of variants with improved photophysical properties.
Reference:
Mickael Lelimousin, Virgile Adam, G. Ulrich Nienhaus, Dominique Bourgeois and Martin J. Field. Photoconversion of the Fluorescent Protein EosFP: A Hybrid Potential Simulation Study Reveals Intersystem Crossings. JACS (2009) 131:16814-23.

How do fluorescent proteins switch?
Text to come
Simulation of the cis to trans isomerization of the chromophore by hula-twist mechanism
3D structure of the chromophoric environment of a green FP in its cis conformation. 2Fo-Fc electron density map is shown, contoured at 1σ
3D structure of the chromophoric environment of a green FP in its trans conformation. 2Fo-Fc electron density map is shown, contoured at 1σ
Various cellular compartments such as the endoplasmic reticulum or the mitochondrial intermembrane space may be considered as "hostile" environments, because particularly oxidizing. This is also the case in the bacterial periplasm, a space of major importance for the understanding of cellular respiration, biofilms formation and antibiotic resistance.
When proteins of interest are fused to fluorescent proteins to allow their microscopic observation, the latter, once secreted into the oxidizing environments, are generally unable to fold correctly and thus fluoresce. There is one notable exception, Superfolder GFP, unfortunately unsuitable for super resolution microscopy.
In this work, combining biochemistry, crystallography and photophysical studies, we realized the rational engineering of Superfolder-GFP in order to make this marker photoswitchable.
To this purpose, we constructed a chimeric protein combining Superfolder-GFP and rsEGFP2, a GFP derivative used for super-resolution in non-oxidizing environments. The result: rsFolder is a new tool is a new tool allowing the observation of oxidizing environments such as the periplasm with resolutions of the order of 70 nm.
The development of rsFolder is currently ongoing in order to obtain new generations of even more efficient markers for biologists and to access otherwise unobservable hostile cell territories in super-resolution.
Reference:
El Khatib, M., Martins, A., Bourgeois, D., Colletier, J.-P. & Adam, V. Rational design of ultrastable and reversibly photoswitchable fluorescent proteins for super-resolution imaging of the bacterial periplasm. Scientific Reports 6, 18459 (2016).
More recently, we have used serial femtosecond crystallography (SFX) at X-ray free electron laser (XFEL) sources to sudy photoswitching. This method exploits intense X-ray pulses to provide a diffraction pattern before radiation damage destroys the protein crystal. The sample is replenished with millions of tiny microcrystals and diffraction data is collected in a serial way. This enables time-resolved studies of proteins in action down to the femtosecond time scale, which means during the photoswitching itself, and to catch structural details of intermediate states.
Time-resolved and static crystallographic experiments on reversibly switchable fluorescent proteins IrisFP and rsEGFP2 in their on and off state were solved by SFX.
The high-quality structures show no signs of X-ray radiation damage and were determined from a very small amount of crystalline sample for IrisFP using grease as an injection medium. rsEGFP2 crystals were injected in liquid medium and allowed to trap intermediate states along the photoreaction pathway from the trans to the cis isomerization of the chromophore. For this project, the IBS DYNAMOP group used the XFEL at SACLA in Japan and LCLS in the USA and teamed up with scientists the beamlines, the Max-Planck Institute in Heidelberg, the Universities of Lille and Rennes and the ESRF in Grenoble.
As a complement to SFX, time-resolved absorption spectroscopy was used to identify intermediate-states during photoswitching.
Together, our data lay a solid ground for ultra-fast time-resolved SFX at XFELs of photoswitchable fluorescent proteins that, beyond their fascinating photochemistry, are of major importance for advanced nanoscopy, such as super-resolution microscopy.
Reference:
Serial Femtosecond Crystallography and Ultrafast Absorption Spectroscopy of the Photoswitchable Fluorescent Protein IrisFP. Colletier JP, Sliwa M, Gallat FX, Sugahara M, Guillon V, Schiro G, Coquelle N, Woodhouse J, Roux L, Gotthard G, Royant A, Uriarte LM, Ruckebusch C, Joti Y, Byrdin M, Mizohata E, Nango E, Tanaka T, Tono K, Yabashi M, Adam V, Cammarata M, Schlichting I, Bourgeois D, Weik M (2016) The Journal of Physical Chemistry Letters: 882-887

Fluorescent proteins that can both change colour and switch
We developed a fluorescent protein called IrisFP [1], which combined for the first time both photoconversion (change of colour) and photoswitching (reversibly commutation between on and off states) properties. Using X-rays form the ESRF synchrotron, we determined the protein’s atomic structure and characterized each of its four states (green on, green off, red on and red off). IrisFP is the first of highly versatile tools that we termed "biphotochromic". Such tools could considerably advance microscopy techniques by genetically fusing them to proteins of interest, scientists would be able to monitor the protein’s movements of photoconverted proteins among non photoconverted ones (pulse-chase microscopy) at single molecule resolution.
Besides microscopy, the development of new fluorescent probes raises exciting prospects for nanotechnology. Potential future applications include the development of high-density mass storage media that exploit changes in the colour of crystals of these proteins, allowing a large amount of information to be stored in a nanometric-sized structure. We have provided the proof of principle for such data storage applications [2] and below are some examples of 3D storage of information (words) within the depth of a single fluorescent protein crystal.
3D data storage in a protein crystal:
With two-photon microscopy, names of institutes have been written onto 3 different layers of a single IrisFP crystal
3D rendering of the previous stack
Thresholding and extraction of the encoded data in the previous stack
A limitation of IrisFP on cellular microscopy is that it is a tetrameric protein. We thus have developed monomeric versions of biphotochromic fluorescent proteins both from the tetrameric IrisFP, called mIrisFP [3], and from the monomeric photoconvertible fluorescent protein Dendra2, called NijiFP [4]. These tools proved to be efficient in two-colour PALM microscopy and performing pulsed-chase single-molecule microscopy.
Green-to red irreversible photoconversion of NijiFP, absorbance (top) and kinetics (bottom)
Reversible photoswitchings of green NijiFP, absorbance (top) and kinetics (bottom)
Reversible photoswitchings of red NijiFP, absorbance (top) and kinetics (bottom)
Reversible in-vivo photoswitchings of green NijiFP fused to actin in a HeLa cell, image (top) and kinetics (bottom)
References:
[1] Virgile Adam, Mickaël Lelimousin, Susan Boehme, Guillaume Desfonds, Karin Nienhaus, Martin J. Field, Joerg Wiedenmann, Sean McSweeney, G. Ulrich Nienhaus & Dominique Bourgeois, "Structural characterization of IrisFP, an optical highlighter undergoing multiple photo-induced transformations” PNAS (2008), 105, 18343-48.
[2] Virgile Adam, Hideaki Mizuno, Alexei Grichine, Jun-ichi Hotta, Yutaka Yamagata, Benjamien Moeyaert, G. Ulrich Nienhaus, Atsushi Miyawaki, Dominique Bourgeois & Johan Hofkens, "Data storage based on photochromic and photoconvertible fluorescent proteins" Journal of Biotechnology (2010), 149, 289-98.
[3] Jörg Wiedenmann, Susan Gayda, Virgile Adam, Franz Oswald, Karin Nienhaus, Dominique Bourgeois & G. Ulrich Nienhaus, "From EosFP to mIrisFP: structure‐based development of advanced photoactivatable marker proteins of the GFP‐family" Journal of Biophotonics (2011), 4, 377-90
[4] Virgile Adam, Benjamien Moeyaert, Charlotte C. David, Hideaki Mizuno, Mickael Lelimousin, Peter Dedecker, Ryoko Ando, Atsushi Miyawaki, Jan Michiels, Yves Engelborghs & Johan Hofkens, "Rational design of photoconvertible and biphotochromic fluorescent proteins for advanced microscopy applications" Chemistry & Biology (2011), 18, 1241-51

How do fluorescent proteins blink?
Fluorescent proteins from the GFP family are remarkable markers for cell imaging. Their weak photostability, however, constitutes their principal disadvantage. If one observes under the microscope a single fluorescent molecule (a fluorescent protein, or an organic dye for example), blinking can be immediately noticed: fluorescence is not constant over time, but alternates quickly and stochastically between bright (fluorescent) and dark (non-fluorescent) states.
In the case of fluorescent proteins, the molecular and structural origin of blinking remains mysterious. Excited states reactions can generate a transient loss of fluorescence, such as intersystem crossing to the triplet state, chromophore protonation, or chromophore isomerization. Another possibility consists in photo-induced electron transfer, which results in the production of a radical species that is unstable and nonfluorescent.
We have provided evidence for such a radical species [1], which was generated by synchrotron X-rays. By combining crystallography, Raman spectroscopy, and absorption and fluorescence spectroscopy, we could show that the radical state is characterized by a severe distortion of the chromophore, which accounts for the loss of fluorescence.
This study showing the structure of a fluorescent proteins in a transient off state for the first time allowed the development of more photostable variants. This highlights the importance of electron transfer reactions in fluorescent proteins.
In actual microscopy conditions, the blinking process results from illumination with visible light and not with X-rays. Using simulations based on a hybrid approach combining quantum mechanics and molecular mechanics (QM/MM) we demonstrated [2] that fluorescent proteins can blink in essentially the same way under illumination with visible light or X-rays.
The chromophore distortion at the origin of the fluorescence intermittencies can be explained by the reversible transfer of a proton from a nearby arginine residue towards the central part (methylene bridge) of the chromophore in a triplet or a radical state. This distortion of the chromophore disrupts transiently its electronic conjugation and hence stops its fluorescence emission. This work is allows the future development of more photostable fluorescent proteins.
Photoconvertible fluorescent proteins used in PhotoActivated Localization Microscopy (PALM) allow counting target proteins one by one directly inside cells. Unfortunately, the accuracy of counting is limited by blinking events. Indeed, a single molecule that blinks can easily be confounded with an ensemble of distinct molecules that appear successively at the same location.
By combining X-ray crystallography, optical spectroscopy and PALM microscopy, we discovered [3] that the orientation of a unique, fully conserved, aminoacid (arginine 66) located next to the chromophore entirely controls and is sufficient to accurately predict the blinking properties of photoconvertible fluorescent proteins.
This research brings new knowledge in fundamental photophysics and opens the door to the rational engineering of variants optimized for quantitative PALM.
References:
[1] Virgile Adam, Philippe Carpentier, Sebastien Violot, Mickaël Lelimousin, Claudine Darnault, G. Ulrich Nienhaus & Dominique Bourgeois, "Structural Basis of X-ray Induced Photobleaching in a Photoactivatable Green Fluorescent Protein”, J. Am. Chem. Soc., (2009), 131:18063–18065
[2] Arijit Roy, Martin J. Field, Virgile Adam and Dominique Bourgeois. The Nature of Transient Dark States in a Photoactivatable Fluorescent Protein . JACS (2011) 133:18586-9
[3] Romain Berardozzi, Virgile Adam, Alexandre Martins & Dominique Bourgeois Arginine 66 Controls Dark-State Formation in Green-to-Red Photoconvertible Fluorescent Proteins Journal of the American Chemical Society 138, 558-565 (2016)

How do fluorescent proteins die?
Fluorescent proteins are widespread markers in cellular imaging, providing a highly flexible toolbox to investigate live cells. Unfortunately, contrary to organic dyes, fluorescent proteins are particularly sensitive to the photobleaching phenomenon, the definitive loss of fluorescence following photo-induced destruction of the chromophore.
Photobleaching is particularly problematic in super-resolution microscopy techniques, which are being rapidly developed today, limiting the resolution that can be achieved. By combining kinetic crystallography, optical and Raman spectroscopy, molecular dynamics simulations, mass spectrometry, and super resolution microscopy, we have investigated the photophysical mechanisms leading to photobleaching of the fluorescent protein IrisFP.
We have shown that depending on the illumination intensity used for the imaging experiment, two completely different photobleaching mechanisms show up.
At low laser intensity, typical of a standard widefield microscopy experiment, an oxygen-dependent mechanism predominates. On the contrary, at high laser intensity, typical of super-resolution microscopy experiments, a redox-dependent mechanism prevails. The first mechanism, which generates reactive oxygen species (ROS) in the cell is thus expected to be more cytotoxic than the second mechanism, which does not generate such species.
Thus, this work suggests in a counterintuitive manner that by increasing laser intensity at constant those, less cellular damages would be created. This hypothesis now needs to be experimentally verified.
Reference:
Chenxi Duan, Virgile Adam, Martin Byrdin, Jacqueline Ridard, Sylvie Kieffer-Jacquinot, Cécile Morlot, Delphine Arcizet, Isabelle Demachy & Dominique Bourgeois Structural Evidence for a Two-Regime Photobleaching Mechanism in a Reversibly Switchable Fluorescent Protein J. Am. Chem. Soc. (2013), 135: 15841−15850